
Pulmonal arterielle Hypertonie
Inhaltsverzeichnis
Was ist eine pulmonal arterielle Hypertonie (PAH)?
Die pulmonal arterielle Hypertonie (PAH) ist eine von 5 Gruppen der pulmonalen Hypertonie, auch Lungenhochruck genannt. PAH ist eine seltene Erkrankung, die durch eine Druck- und Widerstandserhöhung in den Lungengefäßen charakterisiert ist.
Auf Grund unterschiedlicher Ursachen für die Entstehung der Erkrankung, wird die PAH in Untergruppen unterteilt:
- PAH ohne bekannte Ursache (idiopathische PAH)
- erbliche PAH
- PAH in Verbindung mit Arzneimitteln oder Toxinen
- PAH in Verbindung mit anderen Erkrankungen wie Bindegewebserkrankungen, HIV-Infektion, portale Hypertension, angeborenen Herzfehlern oder Schistosomiasis
- PAH mit Zeichen einer venösen/kapillären Beteiligung
- persistierende PH bei Neugeborenen
In den Industrienationen geht man für PAH von einer Inzidenz pro Jahr (Anzahl der Neuerkrankungen pro Jahr) von ca. 6 Fällen pro 1 Million Erwachsener und einer Prävalenz (bestehende Fälle) von 48-55 Fällen pro 1 Million Erwachsener aus.
Entstehung der pulmonal arteriellen Hypertonie
Charakteristisch für eine pulmonal arterielle Hypertonie (PAH) ist eine Druck- und Widerstandserhöhung in den Lungengefäßen. PAH wird hämodynamisch (die Strömungsmechanik des Blutes betreffend) durch
- einen mittleren pulmonal arteriellen Druck (mPAP) > 20 mmHg,
- einen pulmonal arteriellen Wedge-Druck (PAWP) ≤ 15 mmHg und
- einen pulmonal vaskulären Widerstand (PVR) > 2 Wood-Einheiten
definiert.
Diese verschiedenen Parameter können mittels eines Verfahrens, der Rechtsherzkatheteruntersuchung, gemessen werden.
Mehr Informationen zur Untersuchung mittels Rechtsherzkatheter finden Sie im Abschnitt Diagnose der pulmonal arteriellen Hypertonie
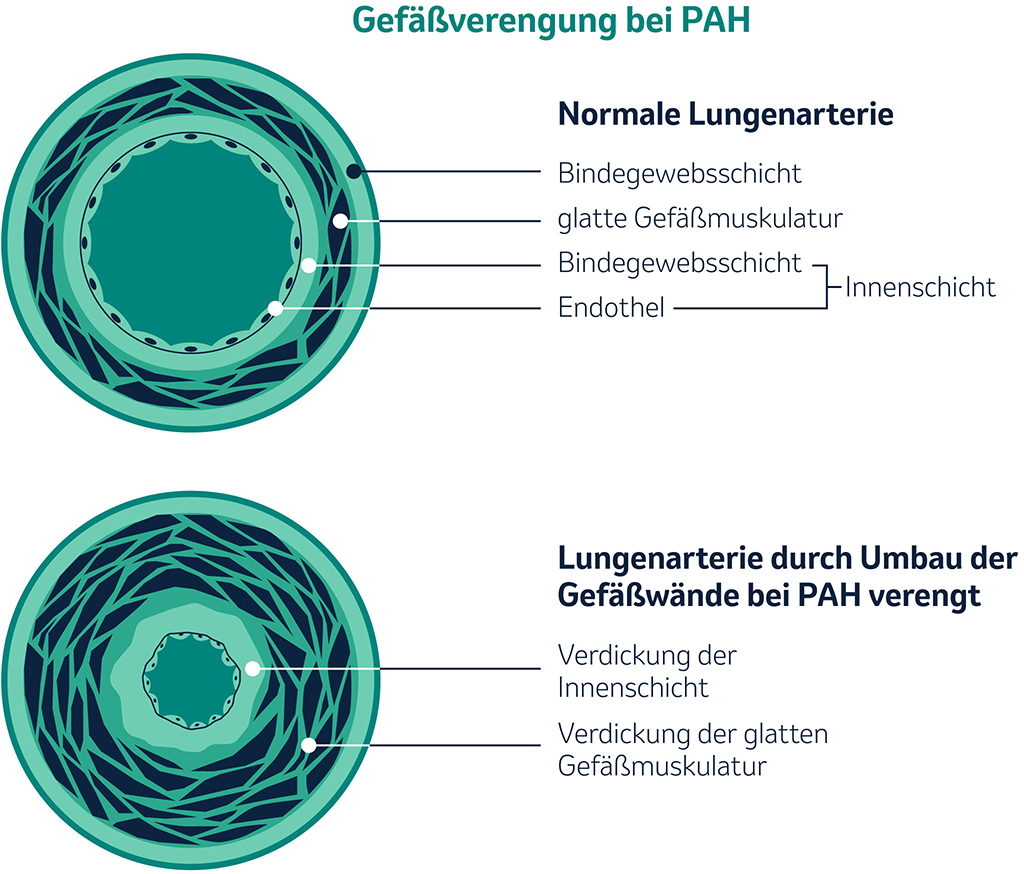
Gefäßverengung bei PAH: Gegenüberstellung einer normalen und einer durch Umbau der Gefäßwände bei PAH verengten Lungenarterie.
Bei PAH verändern sich die weiter vom Herzen entfernt liegenden kleinen Lungengefäße. Durch eine Vermehrung und Vergrößerung der Zellen der Arterienwände, verdicken diese und die Gefäße verengen sich. Dadurch steigt der Widerstand in den Lungenblutgefäßen an, was zu einem höheren Druck in den Lungenarterien führt. Das Herz muss mit einem höheren Druck gegen diesen Widerstand „anpumpen“. Die rechte Herzkammer versucht, mit stärkeren Kontraktionen und dickeren Wänden den erhöhten Druck auszugleichen. Infolgedessen kommt es zu einer Vergrößerung der rechten Herzkammer. Dies kann schlussendlich zum Versagen der rechten Herzkammer führen, was unbehandelt Tod durch Rechtsherzversagen zur Folge hat.
Ursachen der pulmonal arteriellen Hypertonie
Was genau zu einer PAH führt, ist nicht bis ins Detail geklärt. Die Erkrankung kann auf verschiedenen Wegen entstehen und manchmal gibt es keine erkennbare Ursache (idiopathische PAH). Es gibt jedoch eine Reihe von Faktoren, die zur Entstehung der Erkrankung beitragen können, dazu zählen vor allem
- eine familiäre Veranlagung bzw. genetische Faktoren
- Grunderkrankungen, portale Hypertension (erhöhter Blutdruck in der Pfortader der Leber), Bindegewebserkrankungen, HIV-Infektionen, angeborene Herzfehler, Schistosomiasis (eine Parasitenerkrankung)
- manche Medikamente und Drogen, zum Beispiel bestimmte Stimulanzien (Amphetamine), bestimmte Krebsmedikamente, möglicherweise auch Kokain. Bestimmte Appetitzügler, die ebenfalls mit der Entstehung einer PAH in Verbindung gebracht wurden, sind mittlerweile nicht mehr auf dem deutschen Markt erhältlich.

Wichtig: Keiner dieser Faktoren führt definitiv zu einer PAH, sie erhöhen lediglich das Risiko, zu erkranken!
Mit ca. 50-60% der Fälle stellt die idiopathische PAH den häufigsten PAH-Subtyp dar.
Das Durchschnittsalter von PAH-Patient:innen liegt zwischen 43 und 67 Jahren, und Frauen sind in der Regel häufiger von der Krankheit betroffen als Männer (55-81 %). Neuere Daten deuten darauf hin, dass PAH inzwischen häufig bei älteren Patient:innen diagnostiziert wird (d. h. bei Patient:innen im Alter von ≥ 65 Jahren, die oft kardiovaskuläre Begleiterkrankungen haben, was zu einer ausgewogeneren Verteilung zwischen den Geschlechtern führt). Das Durchschnittsalter bei der Diagnosestellung liegt zwischen 50 und 65 Jahren.
Verlauf und Prognose der pulmonal arteriellen Hypertonie
Bei PAH gibt es verschiedene Abstufungen des Schweregrades der Erkrankung, man spricht hier von unterschiedlichen Funktionsklassen. Die funktionelle Klassifikation der Weltgesundheitsorganisation (WHO) ist ein häufig verwendetes Instrument zur Einschätzung der Schwere und Funktionsfähigkeit bei PAH. Die Einteilung in Funktionsklassen (FK) ist ein wertvolles Hilfsmittel, um den Verlauf der PAH zu beurteilen und Entscheidungen bezüglich der Behandlung zu treffen. WHO-FK I oder WHO-FK II sind im Allgemeinen mit einer niedrigeren Ein- und Fünfjahressterblichkeit (Mortalität) assoziiert als WHO-FK III oder WHO-FK IV.
WHO: Funktionelle Klassifikation der pulmonalen Hypertonie (PH)
Funktionelle Klassifikation der PH, modifiziert nach der funktionellen Klassifikation der New York Heart Association gemäß der Weltgesundheitsorganisation 1998.
Patienten mit PH, aber ohne daraus resultierender Einschränkung der körperlichen Aktivität. Gewöhnliche körperliche Aktivität verursacht keine übermäßige Atemnot/Kurzatmigkeit (Dyspnoe) oder Müdigkeit, Brustschmerzen oder Beinahe-Ohnmacht (Synkopen).
Patienten mit PH, die in ihrer körperlichen Aktivität leicht eingeschränkt sind. Sie fühlen sich in Ruhe wohl. Gewöhnliche körperliche Aktivität verursacht übermäßige Atemnot/Kurzatmigkeit oder Müdigkeit, Brustschmerzen oder Beinahe-Synkopen.
Patienten mit PH, die in ihrer körperlichen Aktivität stark eingeschränkt sind. Sie fühlen sich in Ruhe wohl. Weniger als gewöhnliche Aktivität führt zu übermäßiger Atemnot/Kurzatmigkeit oder Müdigkeit, Brustschmerzen oder Beinahe-Synkopen.
Patienten mit PH, die nicht in der Lage sind, eine körperliche Tätigkeit ohne Symptome auszuüben. Diese Patienten weisen Zeichen einer Rechtsherzinsuffizienz auf. Atemnot/Kurzatmigkeit und/oder Müdigkeit können sogar im Ruhezustand auftreten. Die Beschwerden werden durch jedwede körperliche Aktivität verstärkt.
Die Symptome der PAH sind unspezifisch und stehen hauptsächlich im Zusammenhang mit einer fortschreitenden Funktionsstörung der rechten Herzkammer als Folge einer fortschreitenden pulmonalen Gefäßerkrankung.



Das Erscheinungsbild der PAH kann durch Krankheiten, die mit der PAH assoziiert sind, sowie durch Komorbiditäten verändert werden. Man schätzt die Überlebensraten bei PAH nach der Diagnosestellung auf etwa 90,0 % nach 1 Jahr, 69,2 % nach 3 Jahren und 55,3 % nach 5 Jahren.
Das Alter kann für die Überlebensrate genauso eine Rolle spielen wie die Ursache der Erkrankung. Ein Alter unter 65 Jahre war in einer Registerstudie mit einer höheren geschätzten Überlebenswahrscheinlichkeit verbunden als ein Alter über 65 Jahre. Eine HIV-assoziierte PAH und eine PAH assoziiert mit angeborenem Herzfehler zeigten bessere Überlebensraten als andere PAH-Formen. Eine PAH assoziiert mit Bindegewebserkrankungen zeigte im Vergleich die schlechteste Überlebensrate.
Die Prognose bei PAH hängt von verschiedenen Faktoren ab. Dazu zählen
- die Untergruppe der PAH
- der Schweregrad der Erkrankung
- das Vorliegen bzw. das Ausmaß einer Rechtsherzerkrankung
- das Alter
Die Lebenserwartung kann dadurch stark variieren, im Durchschnitt ist sie dank besserer Behandlungsmöglichkeiten in den letzten Jahrzehnten angestiegen.
Aufgrund der unspezifischen Symptome verzögert sich die Diagnose einer PAH häufig. Die Zeit vom Auftreten erster Symptome bis zur finalen Diagnose beträgt meist mehr als 2 Jahre, wobei die Erkrankung zum Zeitpunkt der Diagnose dann häufig schon fortgeschritten ist. Eine frühe Diagnose und ein frühzeitiger Beginn mit der Therapie ist jedoch für die Prognose der Erkrankung von entscheidender Bedeutung.
Diagnose der pulmonal arteriellen Hypertonie
Der erste Verdacht eines Lungenhochdrucks wird aufgrund von klinischen Symptomen gestellt. Da diese Symptome oft sehr unspezifisch sind, werden zur Bestätigung des Verdachtes verschiedene Untersuchungen zunächst beim Kardiologen oder Lungenfacharzt und zur weiteren Abklärung bzw. finalen Diagnosestellung im PH-Zentrum durchgeführt. Durch sie kann auch der Schweregrad der Krankheit festgelegt werden.
Erfahren Sie mehr über die Symptome von pulmonaler Hypertonie
Welche klinischen Untersuchungen werden durchgeführt?
Ein Ultraschall vom Herzen kann Informationen zur Herzstruktur, wie zum Beispiel eine Vergrößerung des Herzens, liefern. In der Regel wird eine Echokardiografie von einem Facharzt für Kardiologie oder Innere Medizin durchgeführt.
Die elektrische Aktivität des Herzmuskels (EKG), die Röntgenaufnahme des Brustkorbs und die Lungenfunktion können auf Grunderkrankungen der PH (z. B. Lungen-, Linksherzerkrankung) hinweisen. Während ein EKG bei einem Hausarzt durchgeführt werden kann, sollten Röntgenthorax und Lungenfunktion in einer Facharztpraxis durchgeführt werden.
Durch die visuelle Darstellung des Herzens können spezielle Befunde (Größe, Morphologie und Funktion des Herzens) erhoben werden, die zur Einschätzung der Schwere der Erkrankung dienen können. Ein MRT wird in einer spezialisierten radiologischen Praxis durchgeführt.
Die nuklearmedizinische Bildgebung der Lungen kann Informationen zu bestimmten Ursachen des Lungenhochdrucks liefern und wird vom Lungenfacharzt durchgeführt.

Bei der Rechtsherzkatheter-Untersuchung wird eine Vene punktiert und ein Katheter eingebracht, der bis zur Lungenarterie geschoben wird, während in verschiedenen Abschnitten die Druckverhältnisse ermittelt werden
Nur durch die Rechtsherzkatheter-Untersuchung wird PAH definitiv diagnostiziert. Während die zuvor aufgeführten Untersuchungen in der Facharzt- bzw. radiologischen Praxis erfolgen, kann eine Rechtsherzkatheter-Untersuchung nur in spezialisierten PH-Zentren durchgeführt werden. Bei dieser Untersuchung kann der pulmonal arterielle Druck (mPAP) genau gemessen werden. Ab einem mPAP >20 mmHg spricht man von einer PAH. Zum Vergleich: Bei gesunden Menschen liegt der mPAP zwischen 8 und 20 mmHg.
Beim 6-Minuten-Gehtest (6MGT) wird die Belastungskapazität bestimmt. Dabei versucht der Patient, in 6 Minuten ohne fremde Hilfe eine möglichst weite Strecke zu gehen. Die zurückgelegte Gehstrecke ist abhängig von Alter, Geschlecht, Gewicht, Größe, Begleiterkrankungen, Bedarf an Sauerstoff, Erlernen und Motivation.
Der 6MGT ist einer von verschiedenen Parametern der bei der Risikostratifizierung im Hinblick auf die geschätzte 1-Jahressterbilichkeit berücksichtig wird.
Eine Spiroergometrie kann mit dem 6-Minuten-Gehtest kombiniert werden. Hierbei wird die Funktionsweise der Lunge in Bezug auf Luftzufuhr und Ausdauer bei körperlicher Belastung auf dem Fahrrad untersucht.
Therapiemöglichkeiten bei pulmonal arterieller Hypertonie
Anhand unterschiedlicher diagnostischer Parameter, wie z. B. den Ergebnissen des 6-Minuten-Gehtests und des Rechtsherzkatheters, wird in den PH-Zentren der Schweregrad der PAH ermittelt und der Krankheitsverlauf eingeschätzt. Auf Grundlage dessen wird eine Therapie eingeleitet.
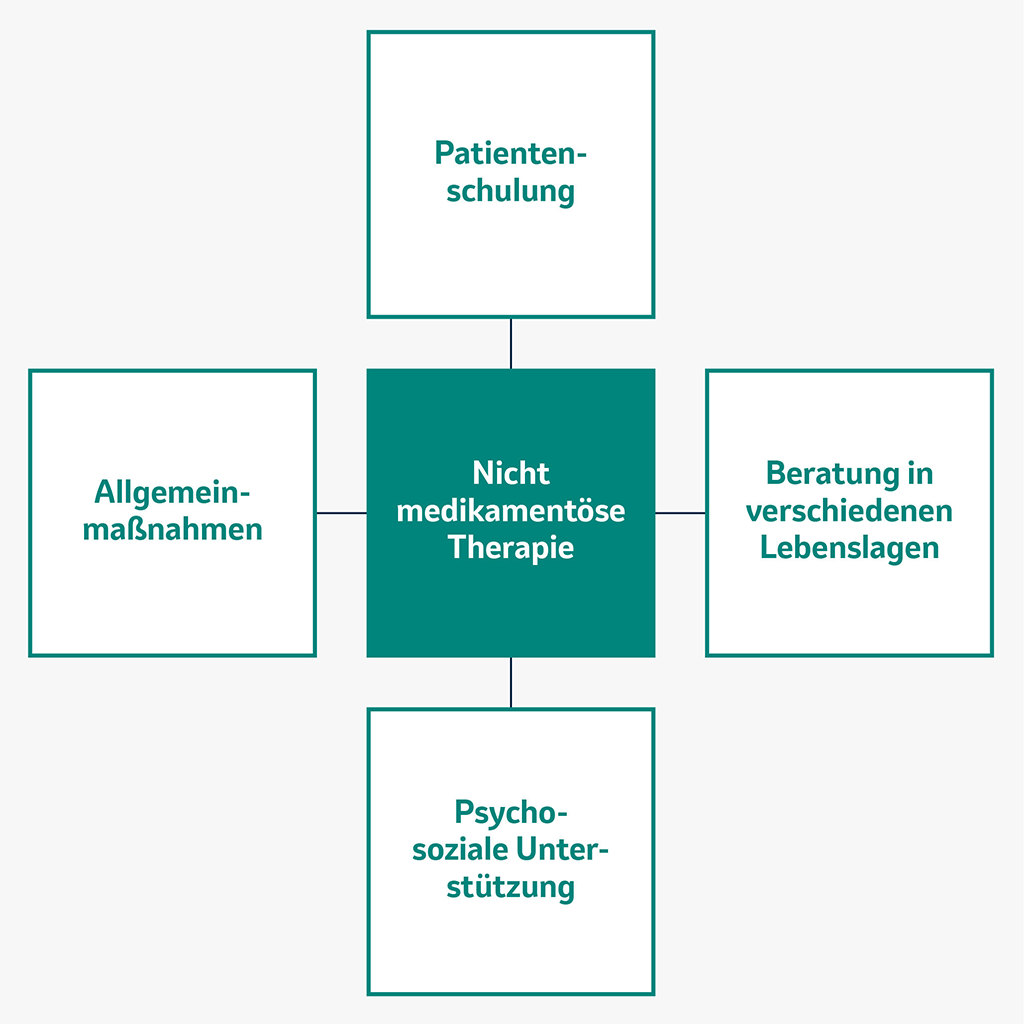
Nicht medikamentöse Maßnahmen bei PAH
Für das Erreichen des Therapieziels – nämlich das Erreichen bzw. Erhalten eines möglichst niedrigen Risikostatus – spielen neben den medikamentösen Therapieoptionen auch Schulungen und Beratungen eine wichtige Rolle.
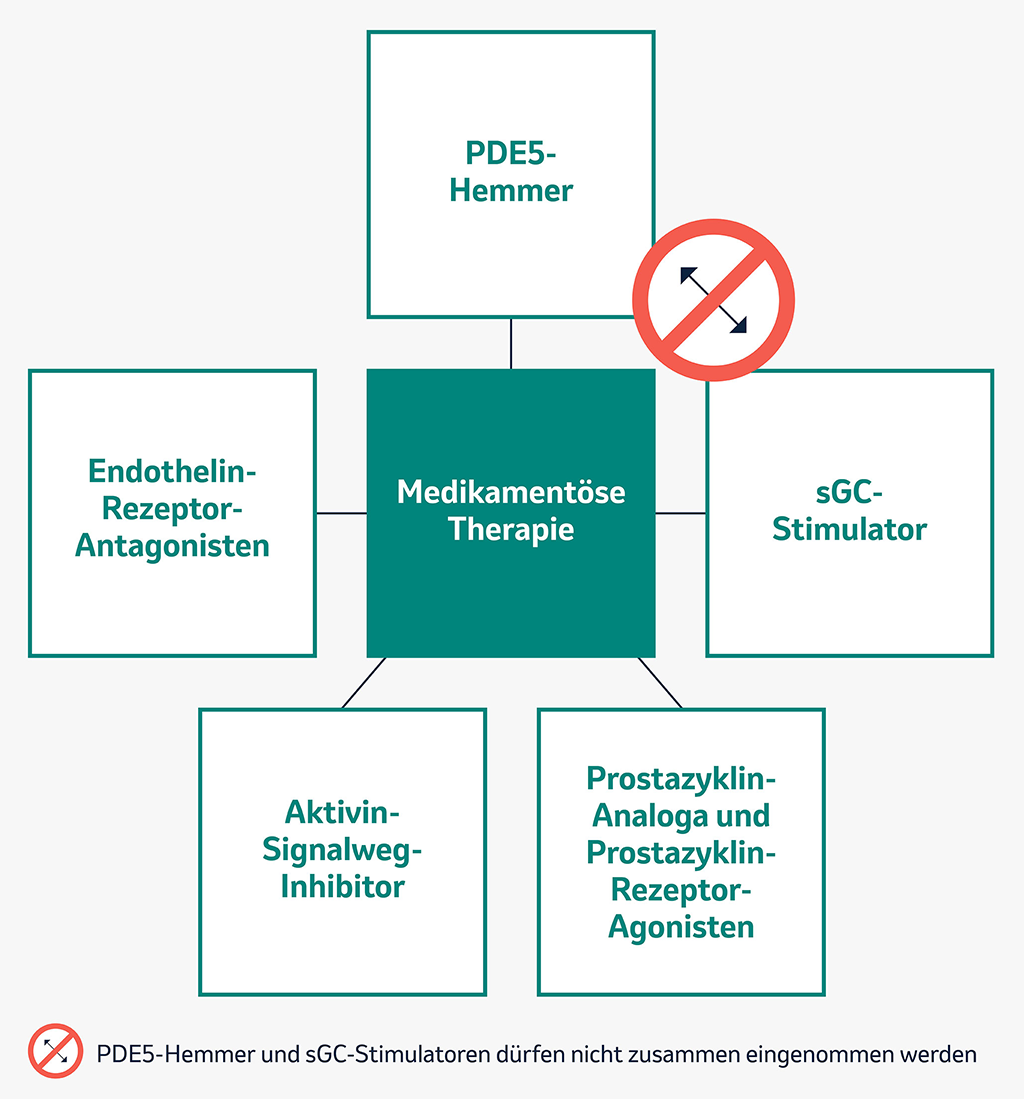
Medikamentöse Therapieoptionen bei PAH
Jedes PAH-Medikament hat einen bestimmten Wirkmechanismus.
Welches Medikament für Sie geeignet ist, entscheidet Ihre Ärztin / Ihr Arzt im PH-Zentrum. Dabei kann es sich um eine Behandlung mit nur einem Medikament oder mehreren Medikamenten, die kombiniert werden, handeln.
Lungentransplantation
Eine Transplantation der Lunge bleibt eine wichtige Behandlungsoption für PAH-Patienten, die auf eine optimierte medikamentöse Therapie nicht ausreichend ansprechen. Die Überweisung zu einem Lungentransplantationszentrum zur weiteren Abklärung sollte bei bestimmten PAH-Patient:innen (z. B. bei lebensbedrohlichen Komplikationen) rasch erfolgen.
Hier finden Sie mehr Informationen zu Selbsthilfegruppen zum Thema PH.
Weiterführende Informationen

Pulmonale Hypertonie
Die pulmonale Hypertonie ist eine Erkrankung mit einer herausfordernden Diagnose. Informationen rund um dieses komplexe Erkrankungsbild lesen Sie hier.

Indikationsbroschüre PAH
Einen Überblick über Lungenhochdruck (pulmonale Hypertonie) im Allgemeinen und über die PAH im Speziellen finden Sie zusammengefasst in dieser Broschüre.

Leben mit PAH
Diese Broschüre beinhaltet viele hilfreiche Infos und Tipps, von Betroffenen für Betroffene sowie deren Angehörige und Freunde.
DE-NON-05728